Clustering Large Applications
clara.RdComputes a "clara" object, a list representing a
clustering of the data into k clusters.
Arguments
- x
data matrix or data frame, each row corresponds to an observation, and each column corresponds to a variable. All variables must be numeric (or logical). Missing values (NAs) are allowed.
- k
integer, the number of clusters. It is required that \(0 < k < n\) where \(n\) is the number of observations (i.e., n =
nrow(x)).- metric
character string specifying the metric to be used for calculating dissimilarities between observations. The currently available options are "euclidean", "manhattan", "jaccard".
Euclidean distances are root sum-of-squares of differences, and manhattan distances are the sum of absolute differences.
- stand
logical, indicating if the measurements in
xare standardized before calculating the dissimilarities. Measurements are standardized for each variable (column), by subtracting the variable's mean value and dividing by the variable's mean absolute deviation.- cluster.only
logical; if true, only the clustering will be computed and returned, see details.
- samples
integer, say \(N\), the number of samples to be drawn from the dataset. The default,
N = 5, is rather small for historical (and now back compatibility) reasons and we recommend to setsamplesan order of magnitude larger.- sampsize
integer, say \(j\), the number of observations in each sample.
sampsizeshould be higher than the number of clusters (k) and at most the number of observations (\(n =\)nrow(x)). While computational effort is proportional to \(j^2\), see note below, it may still be advisable to set \(j = \)sampsizeto a larger value than the (historical) default.- trace
integer indicating a trace level for diagnostic output during the algorithm.
- medoids.x
logical indicating if the medoids should be returned, identically to some rows of the input data
x. IfFALSE,keep.datamust be false as well, and the medoid indices, i.e., row numbers of the medoids will still be returned (i.medcomponent), and the algorithm saves space by needing one copy less ofx.- keep.data
logical indicating if the (scaled if
standis true) data should be kept in the result. Setting this toFALSEsaves memory (and hence time), but disablesclusplot()ing of the result. Usemedoids.x = FALSEto save even more memory.- rngR
logical indicating if R's random number generator should be used instead of the primitive clara()-builtin one. If true, this also means that each call to
clara()returns a different result – though only slightly different in good situations.- pamLike
logical indicating if the “swap” phase (see
pam, in C code) should use the same algorithm aspam(). Note that from Kaufman and Rousseeuw's description this should have been true always, but as the original Fortran code and the subsequent port to C has always contained a small one-letter change (a typo according to Martin Maechler) with respect to PAM, the default,pamLike = FALSEhas been chosen to remain back compatible rather than “PAM compatible”.- correct.d
logical or integer indicating that—only in the case of
NAs present inx—the correct distance computation should be used instead of the wrong formula which has been present in the original Fortran code and been in use up to early 2016.Because the new correct formula is not back compatible, for the time being, a warning is signalled in this case, unless the user explicitly specifies
correct.d.
Value
If cluster.only is false (as by default),
an object of class "clara" representing the clustering. See
clara.object for details.
If cluster.only is true, the result is the "clustering", an
integer vector of length \(n\) with entries from 1:k.
Details
clara (for "euclidean" and "manhattan") is fully described in
chapter 3 of Kaufman and Rousseeuw (1990).
Compared to other partitioning methods such as pam, it can deal with
much larger datasets. Internally, this is achieved by considering
sub-datasets of fixed size (sampsize) such that the time and
storage requirements become linear in \(n\) rather than quadratic.
Each sub-dataset is partitioned into k clusters using the same
algorithm as in pam.
Once k representative objects have been selected from the
sub-dataset, each observation of the entire dataset is assigned
to the nearest medoid.
The mean (equivalent to the sum) of the dissimilarities of the observations to their closest medoid is used as a measure of the quality of the clustering. The sub-dataset for which the mean (or sum) is minimal, is retained. A further analysis is carried out on the final partition.
Each sub-dataset is forced to contain the medoids obtained from the
best sub-dataset until then. Randomly drawn observations are added to
this set until sampsize has been reached.
When cluster.only is true, the result is simply a (possibly
named) integer vector specifying the clustering, i.e.,clara(x,k, cluster.only=TRUE) is the same as clara(x,k)$clustering but computed more efficiently.
Note
By default, the random sampling is implemented with a very
simple scheme (with period \(2^{16} = 65536\)) inside the Fortran
code, independently of R's random number generation, and as a matter
of fact, deterministically. Alternatively, we recommend setting
rngR = TRUE which uses R's random number generators. Then,
clara() results are made reproducible typically by using
set.seed() before calling clara.
The storage requirement of clara computation (for small
k) is about
\(O(n \times p) + O(j^2)\) where
\(j = \code{sampsize}\), and \((n,p) = \code{dim(x)}\).
The CPU computing time (again assuming small k) is about
\(O(n \times p \times j^2 \times N)\), where
\(N = \code{samples}\).
For “small” datasets, the function pam can be used
directly. What can be considered small, is really a function
of available computing power, both memory (RAM) and speed.
Originally (1990), “small” meant less than 100 observations;
in 1997, the authors said “small (say with fewer than 200
observations)”; as of 2006, you can use pam with
several thousand observations.
Author
Kaufman and Rousseeuw (see agnes), originally.
Metric "jaccard": Kamil Kozlowski (@ownedoutcomes.com)
and Kamil Jadeszko.
All arguments from trace on, and most R documentation and all
tests by Martin Maechler.
See also
agnes for background and references;
clara.object, pam,
partition.object, plot.partition.
Examples
## generate 500 objects, divided into 2 clusters.
x <- rbind(cbind(rnorm(200,0,8), rnorm(200,0,8)),
cbind(rnorm(300,50,8), rnorm(300,50,8)))
clarax <- clara(x, 2, samples=50)
clarax
#> Call: clara(x = x, k = 2, samples = 50)
#> Medoids:
#> [,1] [,2]
#> [1,] 0.8963047 0.5684269
#> [2,] 49.2389207 51.5137030
#> Objective function: 10.15147
#> Clustering vector: int [1:500] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 ...
#> Cluster sizes: 200 300
#> Best sample:
#> [1] 10 26 28 34 40 42 46 53 55 63 76 86 115 118 125 140 157 173 175
#> [20] 202 214 262 267 271 272 277 330 347 350 375 382 389 402 413 416 418 434 443
#> [39] 445 449 454 459 470 493
#>
#> Available components:
#> [1] "sample" "medoids" "i.med" "clustering" "objective"
#> [6] "clusinfo" "diss" "call" "silinfo" "data"
clarax$clusinfo
#> size max_diss av_diss isolation
#> [1,] 200 24.00382 10.40779 0.3417825
#> [2,] 300 25.62774 9.98059 0.3649051
## using pamLike=TRUE gives the same (apart from the 'call'):
all.equal(clarax[-8],
clara(x, 2, samples=50, pamLike = TRUE)[-8])
#> [1] TRUE
plot(clarax)
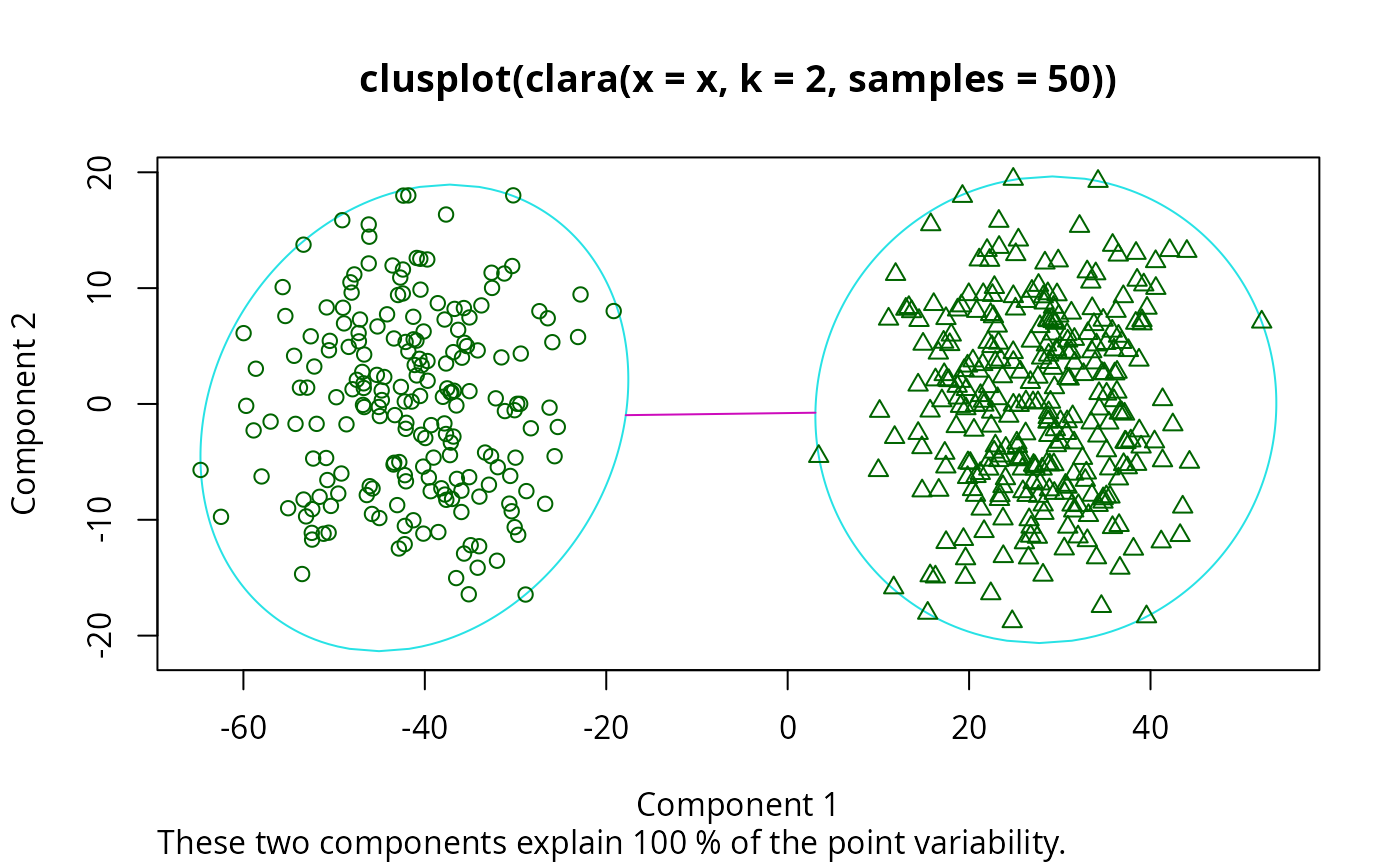
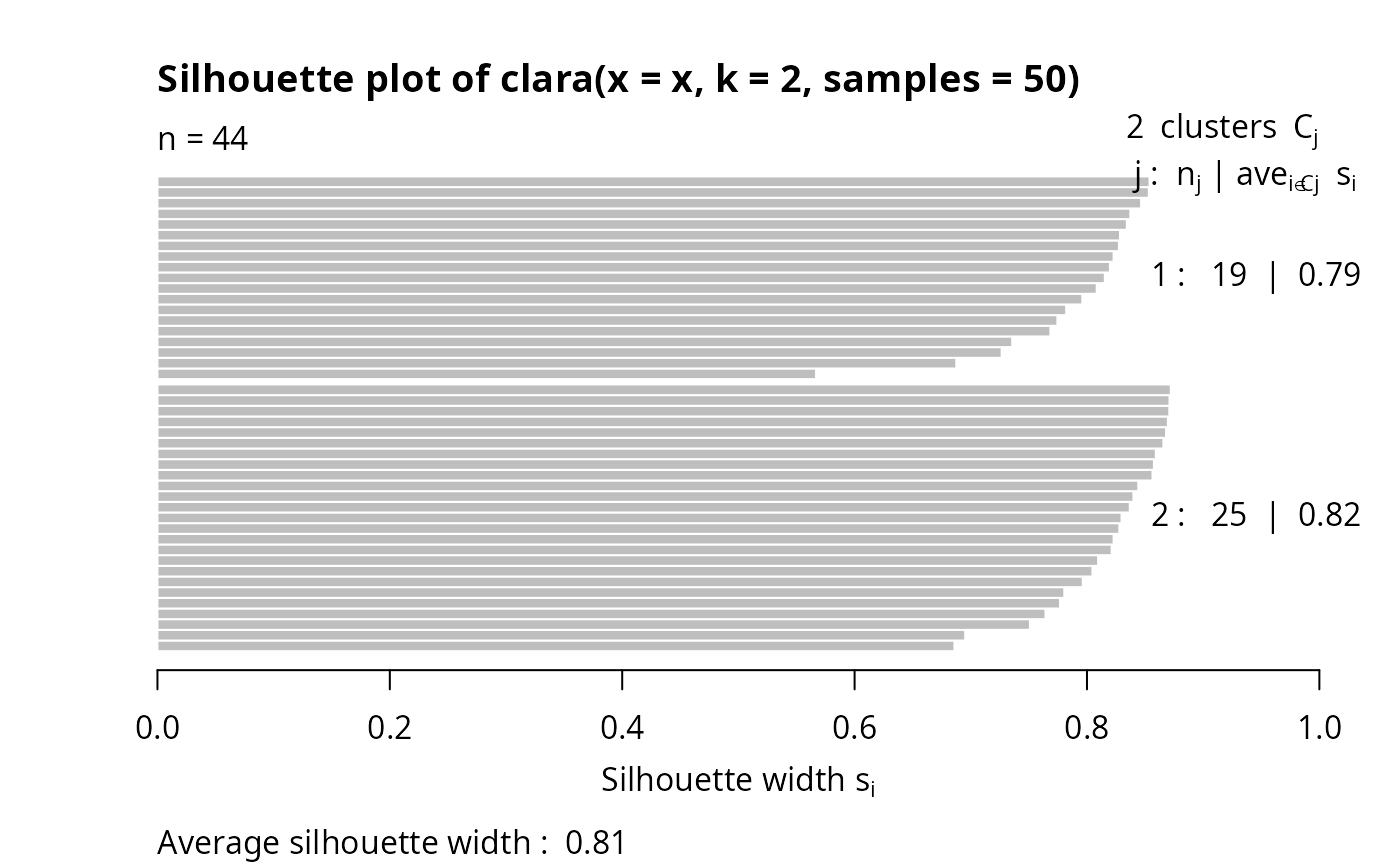 ## cluster.only = TRUE -- save some memory/time :
clclus <- clara(x, 2, samples=50, cluster.only = TRUE)
stopifnot(identical(clclus, clarax$clustering))
## 'xclara' is an artificial data set with 3 clusters of 1000 bivariate
## objects each.
data(xclara)
(clx3 <- clara(xclara, 3))
#> Call: clara(x = xclara, k = 3)
#> Medoids:
#> V1 V2
#> [1,] 5.553391 13.306260
#> [2,] 43.198760 60.360720
#> [3,] 74.591890 -6.969018
#> Objective function: 13.225
#> Clustering vector: int [1:3000] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 ...
#> Cluster sizes: 900 1148 952
#> Best sample:
#> [1] 20 30 46 91 92 169 179 187 209 223 382 450 555 971 1004
#> [16] 1025 1058 1277 1281 1302 1319 1361 1362 1513 1591 1623 1628 1729 1752 1791
#> [31] 1907 1917 1946 2064 2089 2498 2527 2537 2545 2591 2672 2722 2729 2790 2797
#> [46] 2852
#>
#> Available components:
#> [1] "sample" "medoids" "i.med" "clustering" "objective"
#> [6] "clusinfo" "diss" "call" "silinfo" "data"
## "better" number of samples
cl.3 <- clara(xclara, 3, samples=100)
## but that did not change the result here:
stopifnot(cl.3$clustering == clx3$clustering)
## Plot similar to Figure 5 in Struyf et al (1996)
if (FALSE) plot(clx3, ask = TRUE) # \dontrun{}
## cluster.only = TRUE -- save some memory/time :
clclus <- clara(x, 2, samples=50, cluster.only = TRUE)
stopifnot(identical(clclus, clarax$clustering))
## 'xclara' is an artificial data set with 3 clusters of 1000 bivariate
## objects each.
data(xclara)
(clx3 <- clara(xclara, 3))
#> Call: clara(x = xclara, k = 3)
#> Medoids:
#> V1 V2
#> [1,] 5.553391 13.306260
#> [2,] 43.198760 60.360720
#> [3,] 74.591890 -6.969018
#> Objective function: 13.225
#> Clustering vector: int [1:3000] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 ...
#> Cluster sizes: 900 1148 952
#> Best sample:
#> [1] 20 30 46 91 92 169 179 187 209 223 382 450 555 971 1004
#> [16] 1025 1058 1277 1281 1302 1319 1361 1362 1513 1591 1623 1628 1729 1752 1791
#> [31] 1907 1917 1946 2064 2089 2498 2527 2537 2545 2591 2672 2722 2729 2790 2797
#> [46] 2852
#>
#> Available components:
#> [1] "sample" "medoids" "i.med" "clustering" "objective"
#> [6] "clusinfo" "diss" "call" "silinfo" "data"
## "better" number of samples
cl.3 <- clara(xclara, 3, samples=100)
## but that did not change the result here:
stopifnot(cl.3$clustering == clx3$clustering)
## Plot similar to Figure 5 in Struyf et al (1996)
if (FALSE) plot(clx3, ask = TRUE) # \dontrun{}
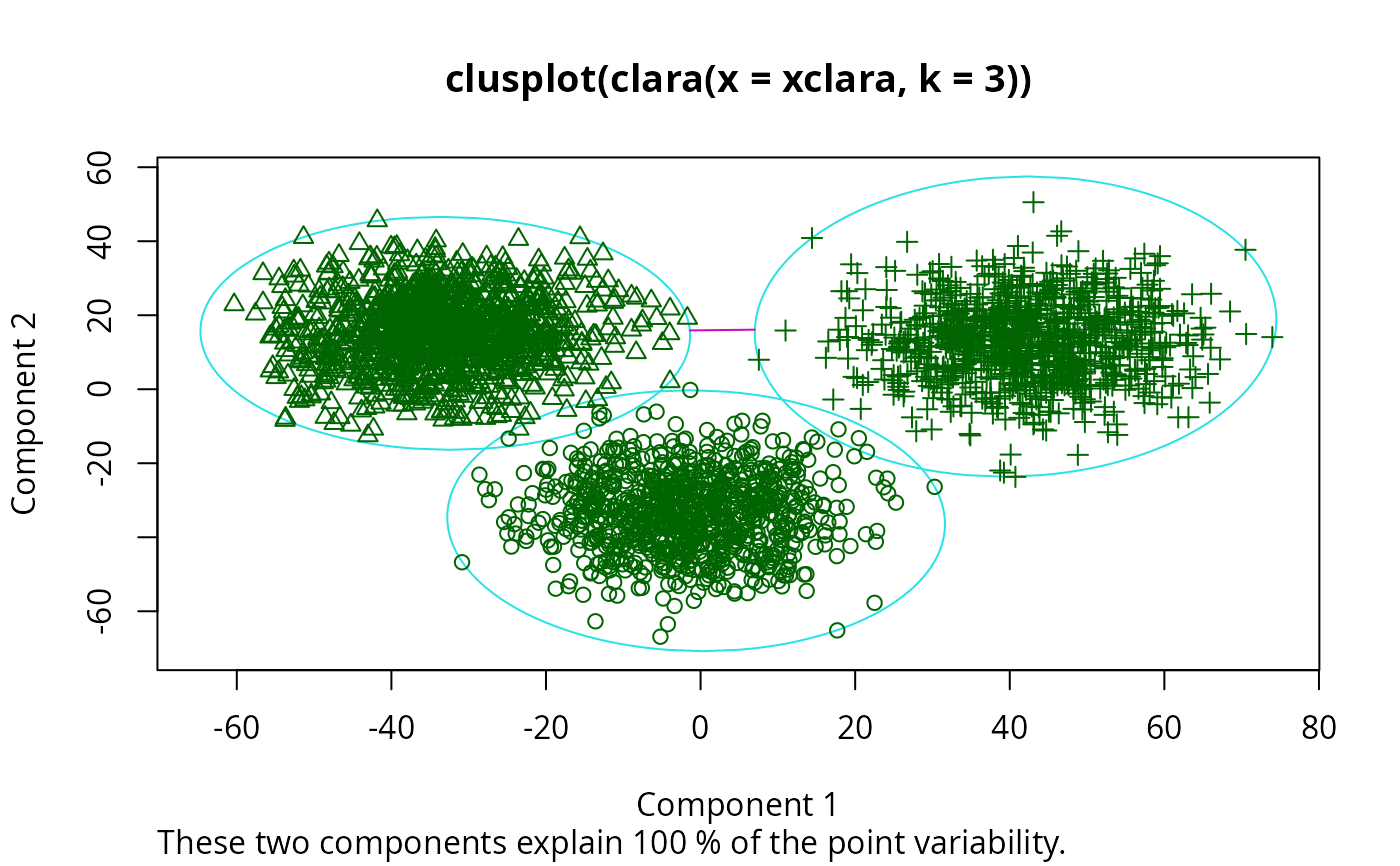
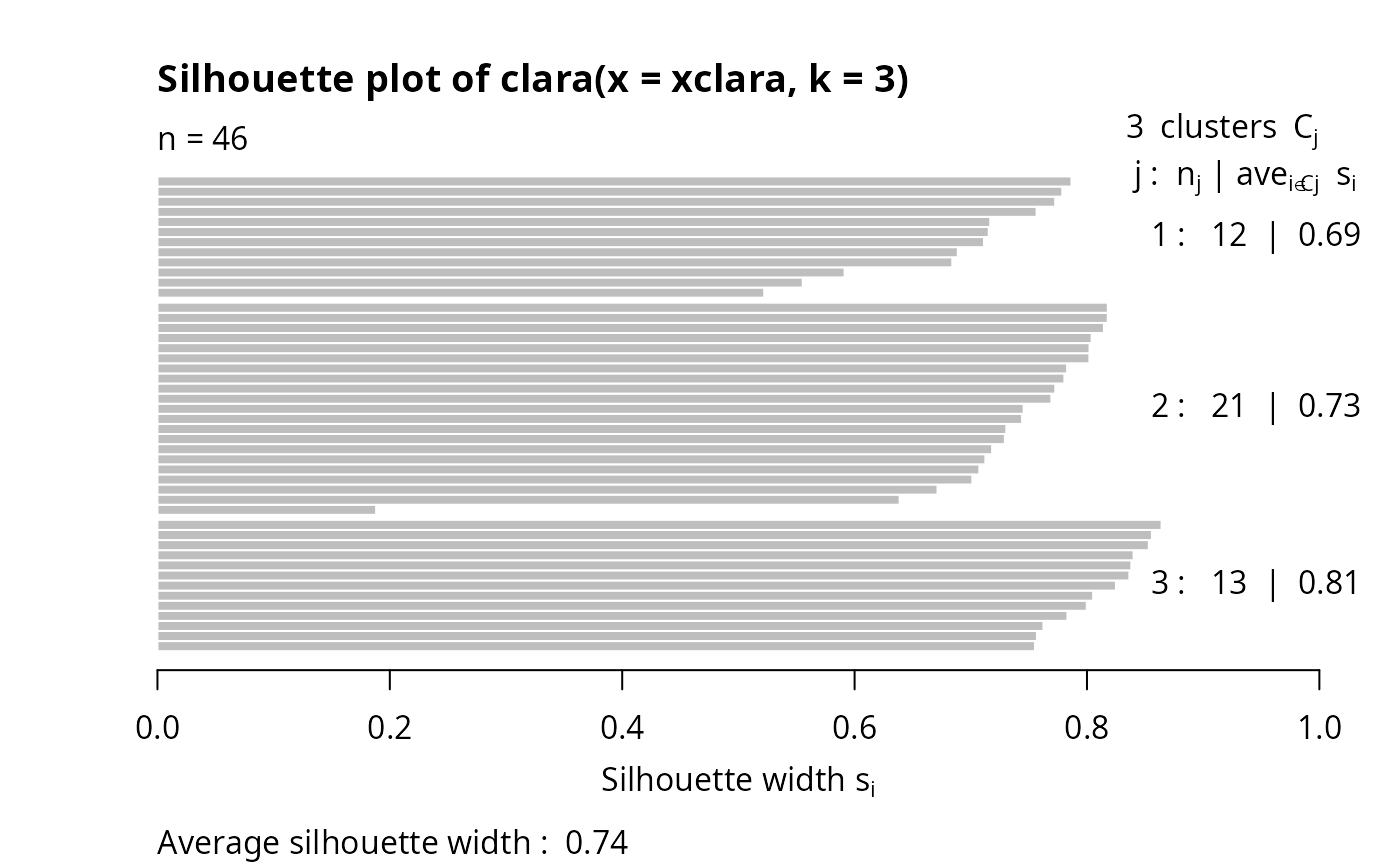 ## Try 100 times *different* random samples -- for reliability:
nSim <- 100
nCl <- 3 # = no.classes
set.seed(421)# (reproducibility)
cl <- matrix(NA,nrow(xclara), nSim)
for(i in 1:nSim)
cl[,i] <- clara(xclara, nCl, medoids.x = FALSE, rngR = TRUE)$clustering
tcl <- apply(cl,1, tabulate, nbins = nCl)
## those that are not always in same cluster (5 out of 3000 for this seed):
(iDoubt <- which(apply(tcl,2, function(n) all(n < nSim))))
#> [1] 30 243 245 309 562 610 708 727 770 1038 1081 1120 1248 1289 1430
#> [16] 1610 1644 1683 1922 2070 2380 2662 2821 2983
if(length(iDoubt)) { # (not for all seeds)
tabD <- tcl[,iDoubt, drop=FALSE]
dimnames(tabD) <- list(cluster = paste(1:nCl), obs = format(iDoubt))
t(tabD) # how many times in which clusters
}
#> cluster
#> obs 1 2 3
#> 30 4 96 0
#> 243 99 0 1
#> 245 91 0 9
#> 309 99 0 1
#> 562 4 0 96
#> 610 82 18 0
#> 708 87 13 0
#> 727 92 0 8
#> 770 2 1 97
#> 1038 81 19 0
#> 1081 44 56 0
#> 1120 12 88 0
#> 1248 22 78 0
#> 1289 5 95 0
#> 1430 1 99 0
#> 1610 57 43 0
#> 1644 24 76 0
#> 1683 1 99 0
#> 1922 13 87 0
#> 2070 2 0 98
#> 2380 4 0 96
#> 2662 4 0 96
#> 2821 8 0 92
#> 2983 2 0 98
## Try 100 times *different* random samples -- for reliability:
nSim <- 100
nCl <- 3 # = no.classes
set.seed(421)# (reproducibility)
cl <- matrix(NA,nrow(xclara), nSim)
for(i in 1:nSim)
cl[,i] <- clara(xclara, nCl, medoids.x = FALSE, rngR = TRUE)$clustering
tcl <- apply(cl,1, tabulate, nbins = nCl)
## those that are not always in same cluster (5 out of 3000 for this seed):
(iDoubt <- which(apply(tcl,2, function(n) all(n < nSim))))
#> [1] 30 243 245 309 562 610 708 727 770 1038 1081 1120 1248 1289 1430
#> [16] 1610 1644 1683 1922 2070 2380 2662 2821 2983
if(length(iDoubt)) { # (not for all seeds)
tabD <- tcl[,iDoubt, drop=FALSE]
dimnames(tabD) <- list(cluster = paste(1:nCl), obs = format(iDoubt))
t(tabD) # how many times in which clusters
}
#> cluster
#> obs 1 2 3
#> 30 4 96 0
#> 243 99 0 1
#> 245 91 0 9
#> 309 99 0 1
#> 562 4 0 96
#> 610 82 18 0
#> 708 87 13 0
#> 727 92 0 8
#> 770 2 1 97
#> 1038 81 19 0
#> 1081 44 56 0
#> 1120 12 88 0
#> 1248 22 78 0
#> 1289 5 95 0
#> 1430 1 99 0
#> 1610 57 43 0
#> 1644 24 76 0
#> 1683 1 99 0
#> 1922 13 87 0
#> 2070 2 0 98
#> 2380 4 0 96
#> 2662 4 0 96
#> 2821 8 0 92
#> 2983 2 0 98